Many chemical reactions occur in condensed phases, and such reactions are used in functional macromolecules such as biomolecules. Yet, how these reactions proceed in confined environments, and how such reaction events trigger functions are often not well understood in detail. In our group, we apply theoretical and computational chemistry to reveal the mechanism of chemical reactions in condensed phases and its role in macromolecular function in molecular details. We develop new theoretical tools to study the dynamics occurring along chemical reactions, and also incorporate machine learning techniques to extract the molecular essence hidden in the complexity of condensed phase events. Collaborations ongoing with experimental groups are also a key to understanding the mechanisms and designing new functional molecules.
Condensed phase reaction dynamics
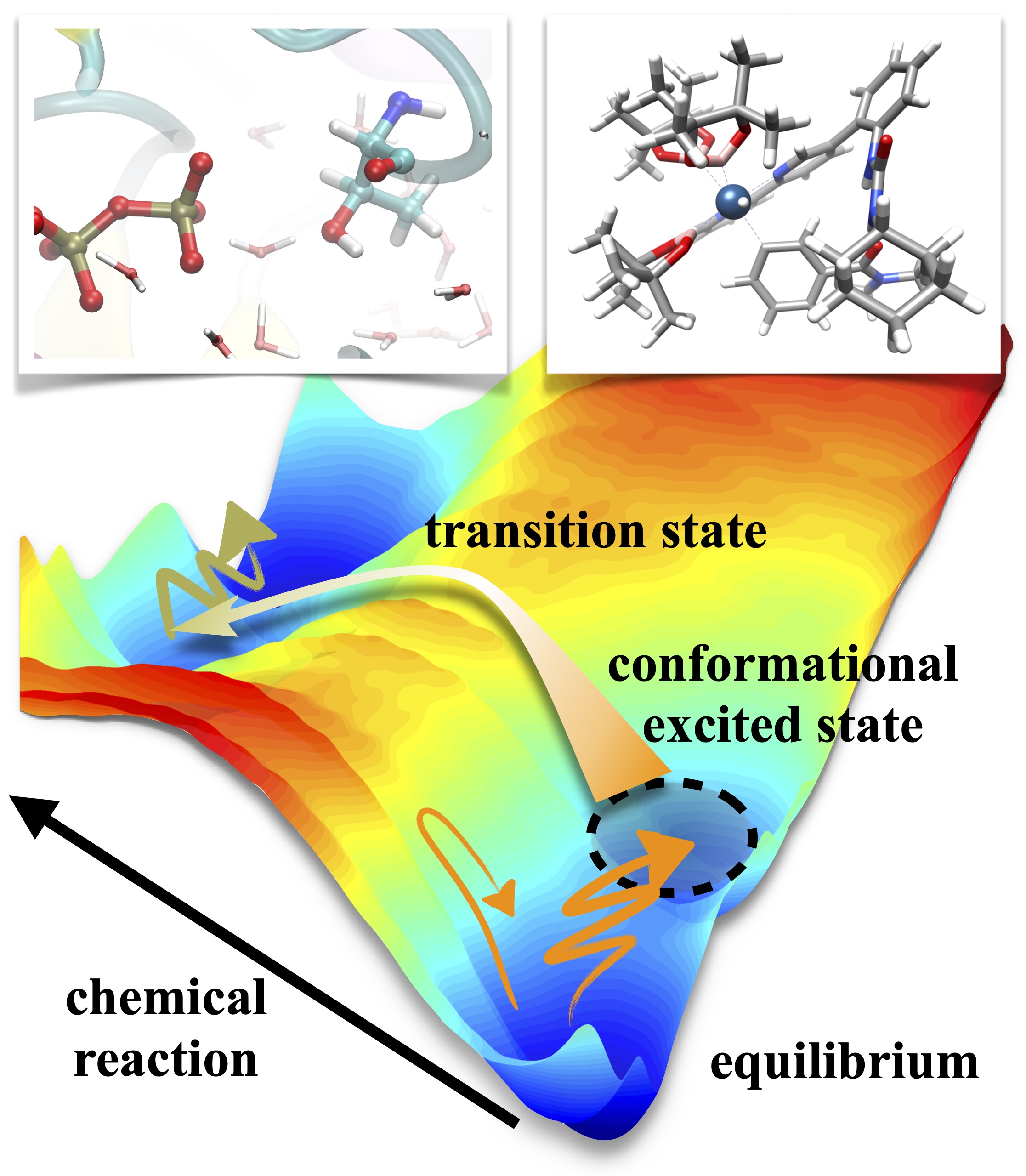
Transition state (a point with highest energy along a reaction path) is essential for understanding chemical reactions, but such point is often not unique in condensed phase. We apply quantum chemistry and molecular dynamics simulation to reveal how reaction actually occurs from dynamic perspective, and use this knowledge to develop principles for catalyst design.
Structure and dynamics of macromolecules
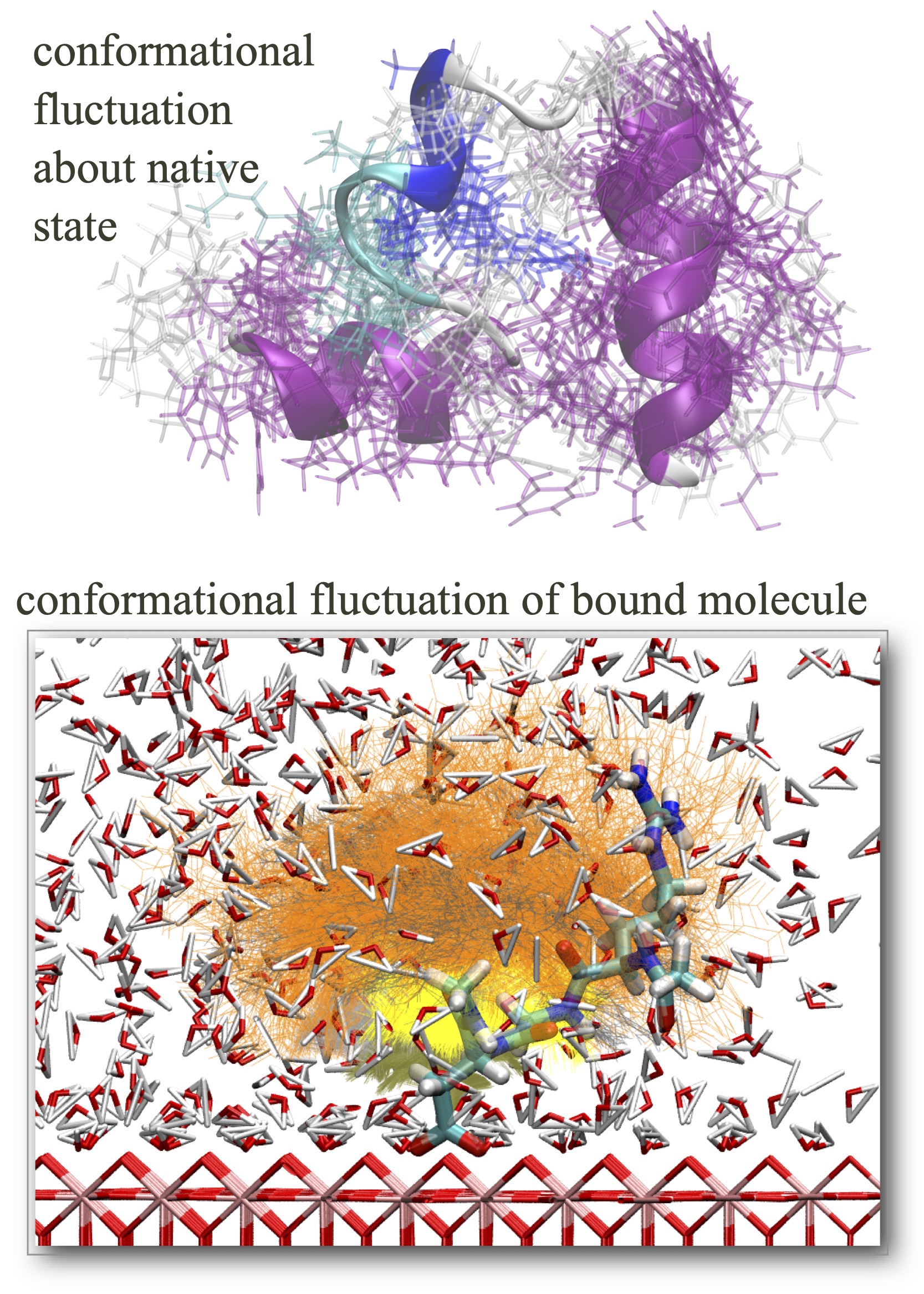
Functional macromolecules, e.g. proteins, are characterized by their unique structures, but in reality they constantly changes its structure. Understanding how conformational transitions occur is thus essential for function. We apply molecular dynamics simulations to reveal the dynamic nature of macromolecules and its role in function.
Development of tools to analyze simulations
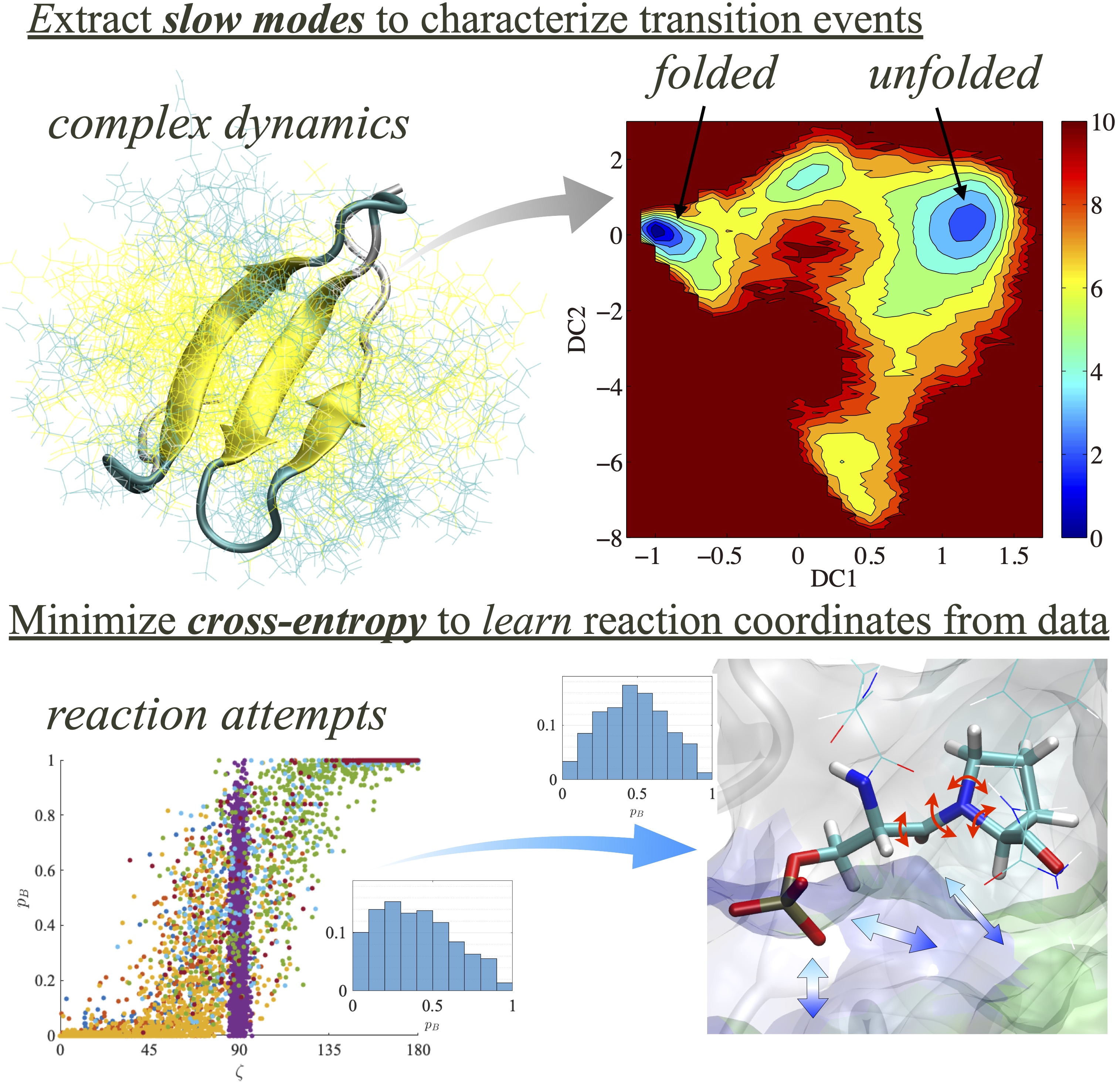
Simulation is a powerful tool to study the molecular events of reaction and conformational dynamics, but due to its high dimensionality, analyzing the data is not trivial. We work on developing theoretical approaches to extract essential information from these trajectories by incorporating machine learning techniques.
Molecular mechanism behind protein functions
Chemical reactions are essential for proteins to function, yet how reactions lead to conformational changes and function are often not clear. We work on the molecular mechanism of behind the sequence of these events. Moreover, we tightly collaborate with experimental groups to understand the mechanism at various levels, and experimentally verify theoretical findings.
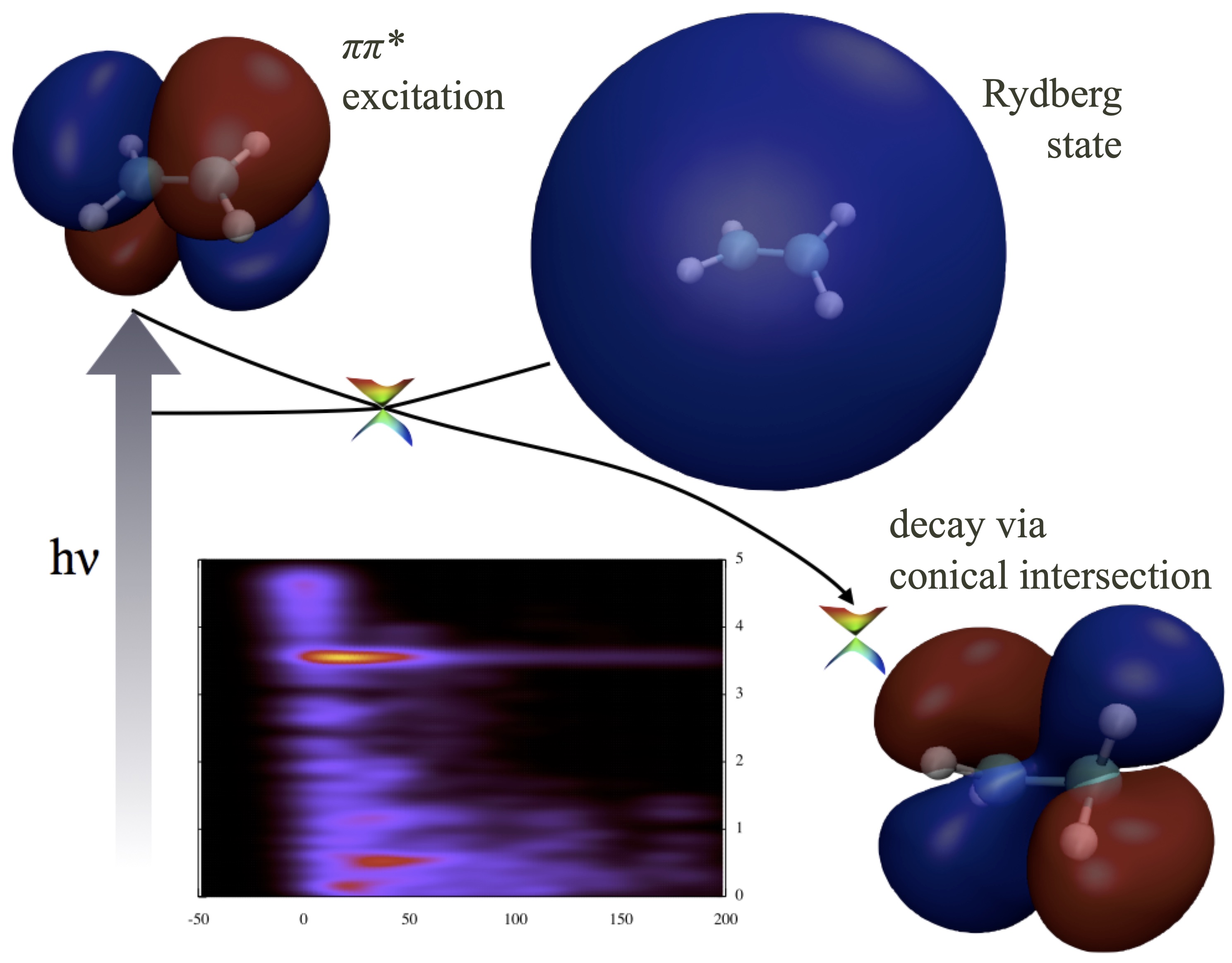
Nonadiabatic dynamics in photochemical reactions
Photoisomerization can convert light energy into mechanical work quickly and efficiently. We study ho such reaction occurs at molecular level using ab initio molecular dynamics simulations. We further develop means to directly compare the simulation to experiments.